
Dalla diagnosi alla terapia
La sindrome da resistenza agli ormoni tiroidei (RTH) è caratterizzata dalla mancata risposta periferica, parziale o totale, dei vari organi bersaglio all’azione degli ormoni tiroidei. Deriva da un difetto dei recettori degli ormoni tiroidei (TR), che possono essere di tipo alfa o beta. In base al tipo di recettore muato si riconoscono allora due forme principali di questa sindrome:
- RTHα: rappresenta una minoranza del totale dei casi
- RTHβ: sindrome rara la cui prevalenza è di circa 1:40.000 nati vivi, con frequenza uguale nei due sessi. Nel 85-90% dei casi si presenta in forma familiare con ereditarietà autosomica dominante.
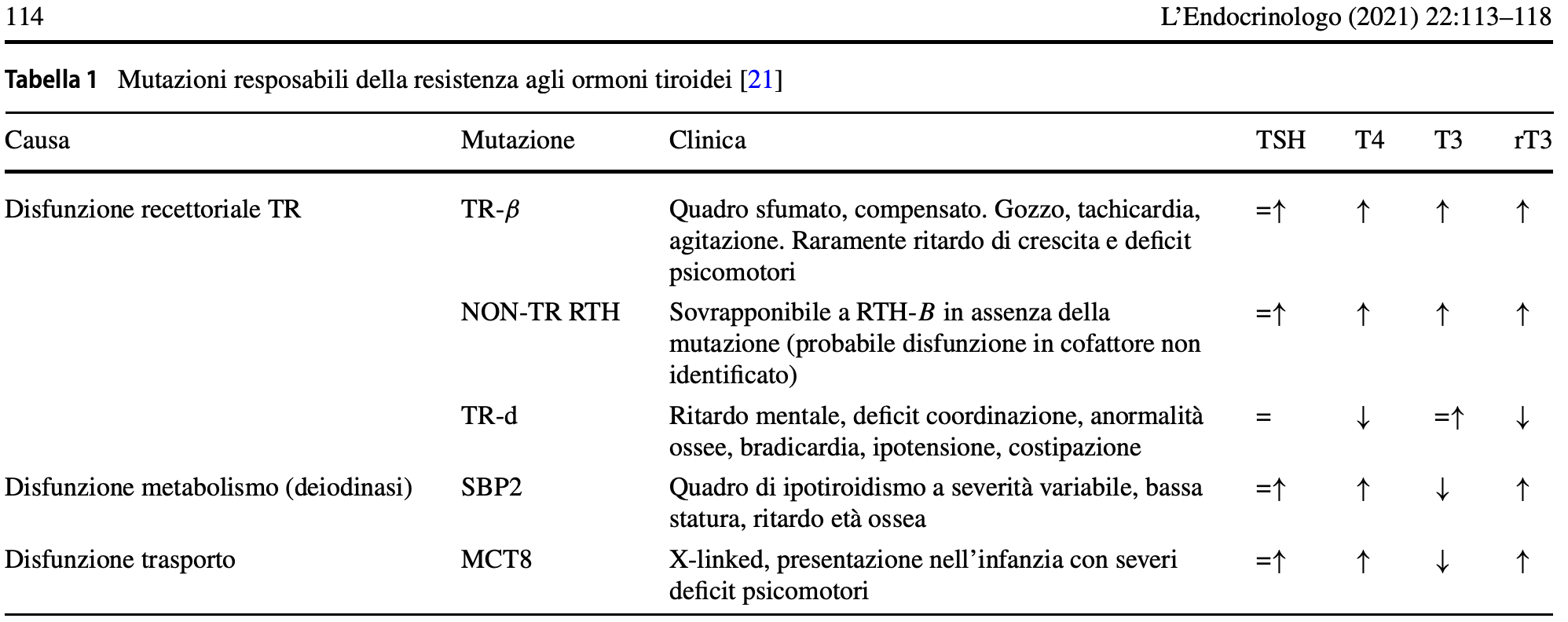
Il tessuto cerebrale esprime principalmente isoforme α ma TR-β-2 è presente in alta concentrazione in alcune aree cerebrali come l’ipotalamo e l’ipofisi, assumendo un ruolo specifico nella regolazione inibitoria operata dalla T3 sulla sintesi di TRH e TSH. Nella RTHβ, l’insensibilità dell’ipofisi agli ormoni tiroidei provoca un aumento compensatorio di T4 e T3 per superare la funzione compromessa del TRβ mutato. La conseguenza è un ipertiroidismo centrale TSH-dipendente. La diversa distribuzione tissutale delle isoforme di TR è responsabile del fenotipo dei pazienti RTHβ, che presentano caratteristiche da tireotossicosi negli organi in cui predomina il TRα (sistema nervoso centrale, muscolo scheletrico, miocardio, osso e intestino) e manifestazioni da ipotiroidismo nei tessuti che esprimono il TRβ (fegato, coclea e retina).
Gli individui affetti da RTH sono in genere eterozigoti; hanno, dunque, un allele THRβ normale e uno mutato, che è sufficiente a produrre il fenotipo clinico. Il TRβ anomalo, infatti, interferisce con la funzione del recettore normale nel fenomeno della dominanza negativa. La completa delezione del gene TRβ descritta in una famiglia ha trasmissione autosomica recessiva; gli individui malati sono omozigoti per la mutazione e mostrano severi segni di ipotiroidismo (ritardo di crescita, deficit neuropsichiatrici e perdita dell’udito).
RTHα è causato da mutazioni eterozigoti dominanti negative nel recettore TRα. I livelli di FT4 sono normali o leggermente ridotti, mentre i livelli di FT3 sono al di sopra del limite superiore, con conseguente rapporto FT4/FT3 ridotto. Questi pazienti mostrano caratteristiche di ipotiroidismo congenito non trattato nel tessuto che esprime il TRα.
Clinica
La maggior parte dei pazienti con RTHβ è asintomatica oppure manifesta sintomi minori di tireotossicosi, come tachicardia, iperidrosi, ansia, irritabilità. I sintomi da tireotossicosi simanifestano nei tessuti che esprimono principalmente il TRα, come il cuore (aumento della frequenza cardiaca), il muscolo scheletrico (aumento del dispendio energetico a riposo) e l’osso (osteopenia/osteoporosi). Alla valutazione clinica, si osservano comunemente tremori fini distali degli arti superiori, gozzo e aumento della frequenza cardiaca. La temperatura corporea e la pressione sanguigna sono normali. Nell’infanzia, si osservano spesso un basso indice di massa corporea (BMI) e uno sviluppo inadeguato, mentre la crescita lineare non è influenzata.
Diagnosi
Questa secrezione inappropriata di TSH è il segno distintivo dell’ipertiroidismo centrale (secondario: ormoni tiroidei alti con TSH o non adeguatamente soppresso), una condizione rara che può essere causata da RTHβ ma anche da un adenoma ipofisario secernente TSH (TSH-oma). Il TSH-oma è la forma più rara di adenoma ipofisario (incidenza annuale di circa 1-2/milione).
I sintomi dei pazienti RTHβ possono essere fuorvianti e sovrapponibili a quelli dei pazienti con TSH-oma. Anche se in quest’ultimo caso la sensibilità agli ormoni tiroidei è conservata e la lenta progressione dell’ipertiroidismo può spiegare la mancanza di manifestazioni gravi.
Se si ha di fronte un paziente con ormoni tiroidei elevati e TSH alto o non adeguatamente soppresso, per cercare di fare diagnosi differenziale e porre un adeguato sospetto clinico, si deve procedere per step:
- Per prima cosa va esclusa un’interferenza di laboratorio, chiedendo una seconda misurazione degli ormoni tiroidei, possibilmente in un centro differente.
- Test dinamici: esplorano l’integrità dell’asse ipotalamo-ipofisi-tiroide e si basano sul presupposto che nella RTHβ, la risposta all’ormone esogeno di rilascio della tireotropina (TRH) sia conservata, mentre la ridotta sensibilità dell’ipofisi può essere superata da alte dosi di ormoni tiroidei. Al contrario, nel TSH-oma la secrezione autonoma di TSH da parte dell’adenoma non può essere regolata da TRH né controregolata dagli ormoni tiroidei. Il test al TRH (iniezione ev di 200 μg di TRH con misurazione del TSH al basale e dopo 20′, 30′, 60′ 90′ minuti) provoca un aumento significativo dei livelli di TSH nei soggetti normali e in quelli con RTHβ, mentre non si osservano cambiamenti significativi nel TSH-oma (tuttavia quasi il 20% del TSH-oma può rispondere paradossalmente al TRH). Il test di soppressione con T3 richiede la somministrazione di T3 per 10 giorni (dosi fisse di 80-100 μg/giorno o dosi crescenti, iniziando con 50 e raddoppiando ogni 3 giorni fino a 200 μg/giorno). Una soppressione completa del TSH dopo la somministrazione di T3 esclude un TSH-oma e indirizza verso RTHβ. Questo test può essere associato a un test TRH il giorno 10. Il test di soppressione con T3 è controindicato nei pazienti con gravi disturbi cardiaci, aritmie e negli anziani dato il rischio di esacerbazione dei problemi cardiaci sottostanti.
- Analisi genetica: fornisce una diagnosi definitiva nell’80-90% dei casi. Le varianti della mutazione di TRβ sono tipicamente distribuite in tre regioni “hot spot” del dominio di legame del ligando. I recettori mutati perdono la loro capacità di modulare l’espressione genica nei tessuti bersaglio e inibiscono l’attività dei recettori wild-type (la cosiddetta dominanza negativa che spiega l’ereditarietà atutosomica dominante della malattia). Nel 10-20% dei casi con un fenotipo biochimico e clinico di RTHβ, nessuna mutazione può essere identificata nel gene THRB e questa condizione è definita come “non-TR – RTH”.
- RM ipofisaria: mostra una ghiandola normale per dimensioni, segnale e captazione del contrasto. Tuttavia, l’imaging deve essere interpretato con cautela poiché le piccole lesioni ipofisarie sono comuni nella popolazione generale e nella RTHβ. Queste lesioni sono considerate “incidentomi ipofisari” non associati a disfunzione ipotalamo-ipofisi. Al contrario, i piccoli TSH-omi possono non essere visualizzati alla risonanza. La scintigrafia tiroidea non è utile perché sia i TSH-omi che la RTHβ danno un ipertiroidismo TSH-mediato che porta ad un aumento dell’assorbimento del pertecnetato in entrambe le condizioni.

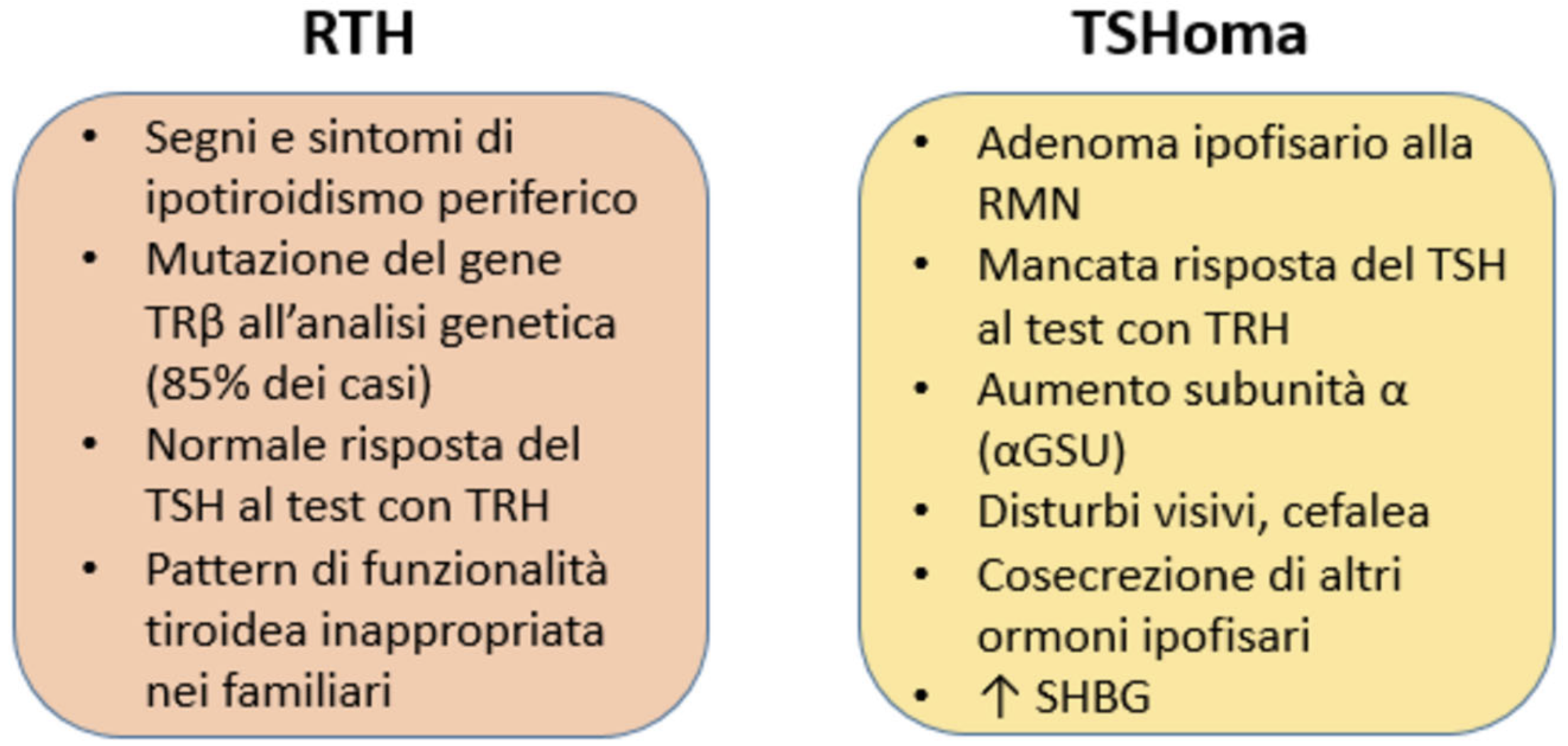
Il telopeptide carbossi-terminale del collagene di tipo I (ICTP) e la globulina legante gli ormoni sessuali (SHBG) sono i marcatori più utilizzati per valutare la sensibilità agli ormoni tiroidei dei tessuti bersaglio (rispettivamente fegato e ossa) e dovrebbero essere normali o ridotti nella RTHβ e aumentati nei pazienti ipertiroidei, compresi quelli con TSH-oma. La SHBG è aumentata in chi utilizza contraccettivi orali. La sensibilità e la specificità di questi marcatori migliorano se valutate durante un test di soppressione T3. La determinazione della subunità alfa degli ormoni glicoproteinici (α-GSU) è di utilità limitata poiché i livelli possono essere normali nei pazienti con piccoli TSH-omi.


Altre indagini strumentali
- Ecografia tiroidea: dovrebbe essere effettuata periodicamente per monitorare eventuali noduli
- Ecocardiogramma: indicato alla diagnosi, poiché è stata riportato un aumento della frequenza di disfunzioni valvolari
- Holter ECG 24h: utile se si sospetta una fibrillazione atriale asintomatica
- MOC DXA: dal momento che RTHβ si associa a osteopenia e osteoporosi
Terapia
L’obiettivo del trattamento è il sollievo dei sintomi, poiché non sono disponibili trattamenti in grado di correggere questi difetti molecolari.
Fortunatamente, nella maggior parte dei pazienti il trattamento non è necessario, in quanto l’iposensitività all’ormone tiroideo è compensata da un’aumentata produzione.
I beta-bloccanti (bisoprololo e atenololo) controllano bene la tachicardia.
Quando i β-bloccanti sono controindicati o persistono sintomi tossici tireotossici, una riduzione dei livelli degli ormoni tiroidei può essere utile. Ciò non può essere ottenuto utilizzando farmaci antitiroidei, perché il conseguente aumento del TSH provoca un aumento del gozzo e iperplasia pituitaria. Al contrario, la somministrazione dell’acido triiodotiroacetico (TRIAC) è il trattamento di scelta in questi casi. TRIAC è anche utile nei bambini RTHβ con ADHD. TRIAC (intervallo terapeutico: 1,4-2,8 mg/die) riduce la secrezione di TSH attraverso il meccanismo di feed-back e la sintesi degli ormoni tiroidei; inoltre, i suoi effetti sono più deboli di quelli di T3, riducendo così le manifestazioni tireotossiche. Durante il trattamento TSH e FT4 sono misurati per l’aggiustamento della dose, mentre FT3 è inaffidabile poiché TRIAC reagisce con T3 nella maggior parte dei saggi immunoistochimici.
Come può essere gestito un ipotiroidismo primario acquisito in un paziente affetto da RTHβ?
Dosi soprafisiologiche di levotiroxina sono necessarie nei pazienti con RTHβ ipotiroidismo a causa di una malattia autoimmune della tiroide associata o dopo l’ablazione tiroidea per una mancata diagnosi di RTHβ. Il trattamento sostitutivo della levotiroxina richiede un attento monitoraggio, valutando non solo il TSH, ma anche i marcatori periferici dell’azione degli ormoni tiroidei.
La chirurgia tiroidea è indicata nei pazienti con RTHβ?
La chirurgia deve essere riservata a pazienti con neoplasie maligne o gozzi grandi che comprimono le vie aeree. Il trattamento con levotiroxina spesso non riesce a mantenere il TSH nell’intervallo preoperatorio a causa della comparsa di sintomi tireotossici, quindi nei pazienti che necessitano di un trattamento TSH-soppressivo (ad esempio, cancro tiroideo differenziato), la levotiroxina può essere combinata con TRIAC.
Lobectomia/tiroidectomia subtotale dovrebbe essere evitata, poiché il gozzo recidiva comunemente, con alterazioni noduolari e asimmetrie grossolane, che richiedono un ulteriore intervento chirurgico o radioiodio.
FONTI
Practical Clinical Endocrinology – Peter Igaz – Springer 2021
Colombo, Paolo & Ariano, Salvatore & Lania, Andrea. (2021). Sindrome da resistenza agli ormoni tiroidei: dalla genetica alla gestione clinica. L’Endocrinologo. 22. 10.1007/s40619-021-00836-1.

Lascia un commento