
UN NUOVO FARMACO CONTRO L’OSTEOPOROSI

Il 29 agosto 2022 è stata aggiornata la Nota 79, tramite cui il Servizio Sanitario Nazionale regola il trattamento per l’osteoporosi e la rimborsabilità dei farmaci utilizzabili, suddividendoli in prima, seconda o terza scelta, a seconda delle condizioni cliniche del paziente. L’esigenza di questa modifica nasce sostanziamente dalla volontà di introdurre in nota un nuovo farmaco per combattere l’osteoporosi, il Rososozumab.
ROMOSOZUMAB: DI CHE SI TRATTA?
Romosozumab è un anticorpo monoclonale umanizzato (IgG2) che si lega alla sclerostina (una glicoproteina prodotta principalmente dagli osteociti ma è espressa anche in altri tessuti, con effetti anti-anabolizzanti sulla formazione ossea, cioè in grado di bloccare la “costruzione” di nuova matrice) inibendola, aumentando di conseguenza la formazione ossea a seguito dell’attivazione delle cellule del rivestimento osseo, aumentando la produzione di matrice ossea da parte degli osteoblasti, e il reclutamento di cellule osteoprogenitrici. Inoltre, romosozumab altera l’espressione dei mediatori osteoclastici, riducendo pertanto il riassorbimento osseo. Complessivamente, questo duplice effetto di aumento della formazione ossea e riduzione del riassorbimento osseo comporta rapidi aumenti nella massa ossea trabecolare e corticale, miglioramenti nella struttura e nella resistenza ossea.
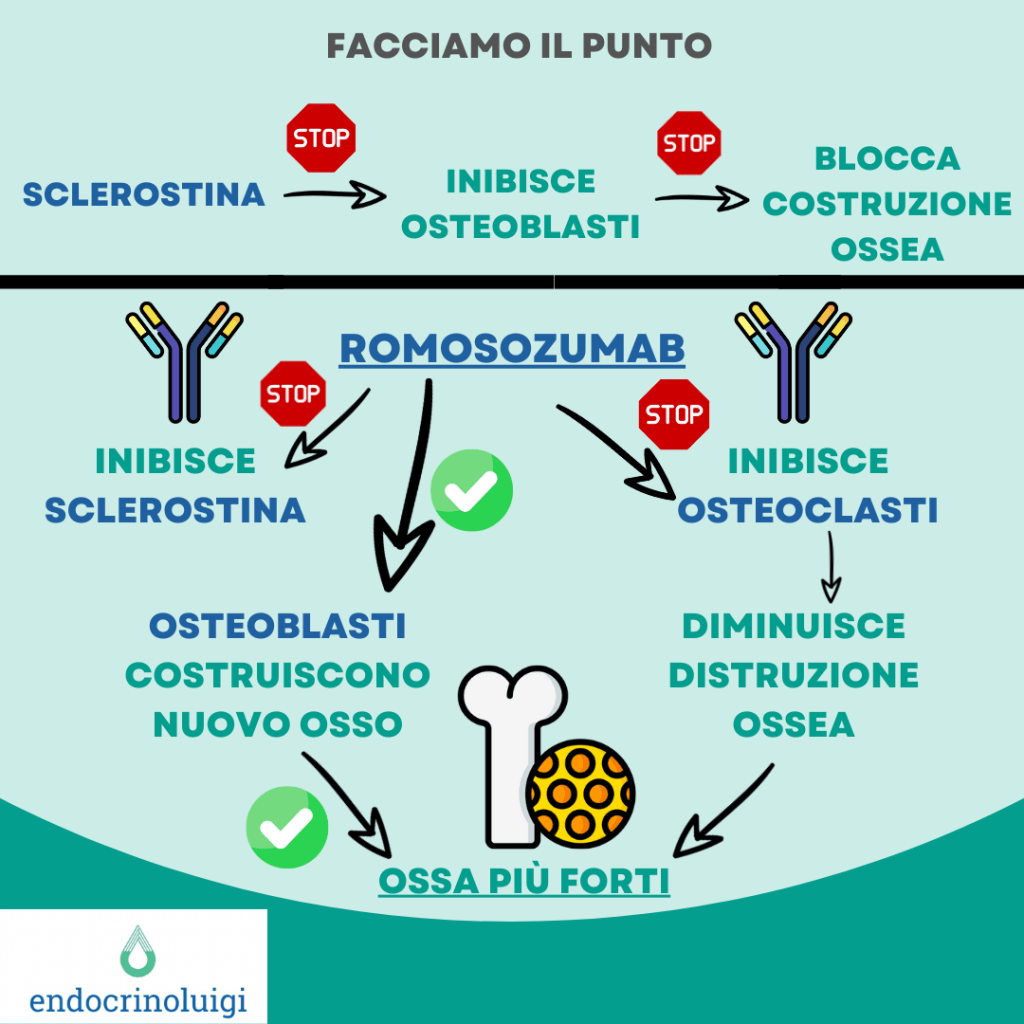
essenziale per la stabilizzazione della β-catenina, un regolatore della trascrizione genica che promuove
l’osteogenesi e inibisce l’osteoclastogenesi. ROMO riattiva la via Wnt, favorendo la formazione ossea e
contemporaneamente inibendo il riassorbimento osseo (azione duplice), contribuendo così al “rafforzamento”
del tessuto osseo.
POSOLOGIA E MODALITA’ DI SOMMINISTRAZIONE
Lo schema posologico prevede per il romosozumab (EVENITY 105 mg soluzione iniettabile) la somministrazione di 210 mg sottocute (somministrati come due iniezioni sottocutanee da 105 mg ciascuna) una volta al mese per 12 mesi. Si può somministrare nell’addome, nella coscia o nella parte superiore del braccio. La seconda iniezione deve essere effettuata immediatamente dopo la prima, ma in una diversa sede.
Se la dose di romosozumab viene dimenticata, occorre somministrarla non appena possibile. In seguito, la dose successiva di romosozumab non deve essere somministrata prima di un mese dopo l’ultima dose.
Studi di fase Il hanno stabilito in 12 mesi la durata massima consigliabile per il trattamento, non ottenendosi per durate più prolungate significativi incrementi di mineralizzazione. Al termine di tale periodo viene raccomandato un trattamento con un farmaco anti-riassorbitivo (bisfosfonati o denosumab) al fine di mantenere i risultati ottenuti e ridurre il rischio di frattura.
Romosozumab incrementa la BMD più di teriparatide, denosumab e alendronato. Un anno di trattamento con ROMO produce miglioramenti della BMD comparabili a sette anni di denosumab. Tuttavia, gli effetti dipendono dal tipo e dalla durata del trattamento precedente (ridotti nei trattati in precedenza con anti-riassorbitivi).
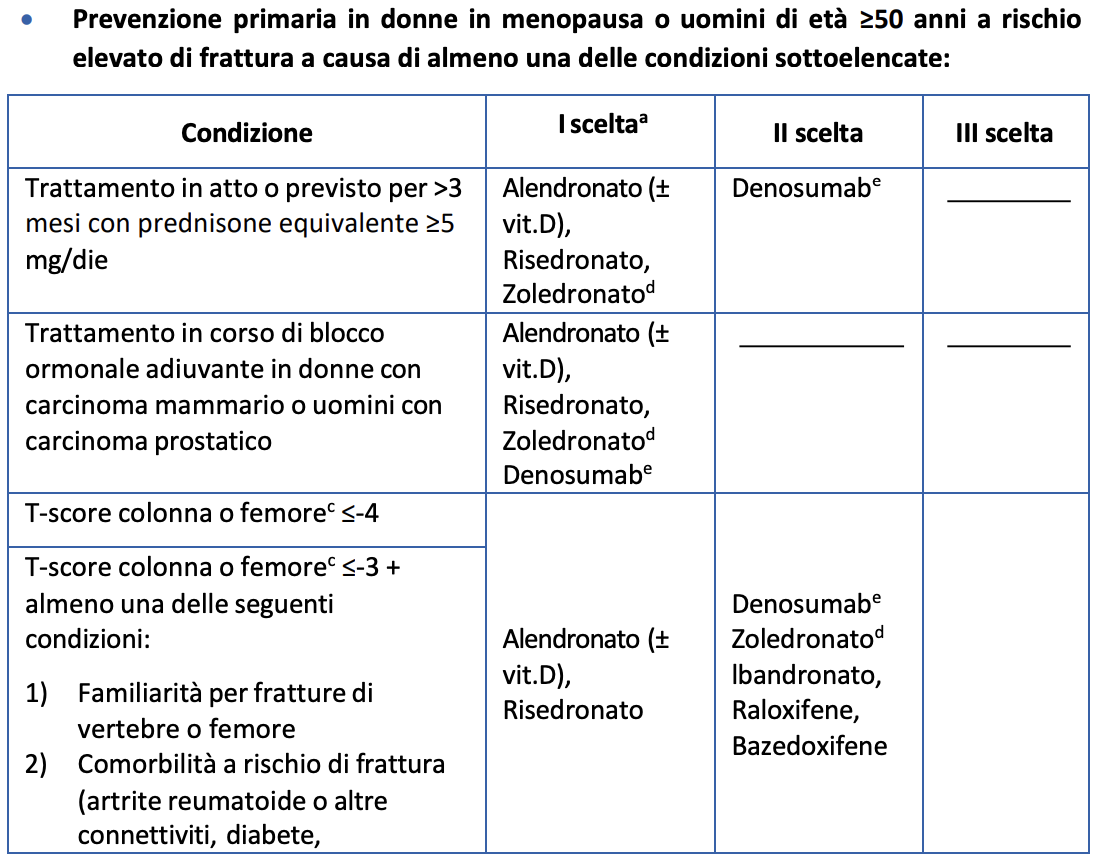
A CHI PRESCRIVERLO?
L’analisi della letteratura disponibile autorizza al momento la prescrizione di romosozumab esclusivamente a pazienti di sesso femminile che presentino le seguenti caratteristiche:
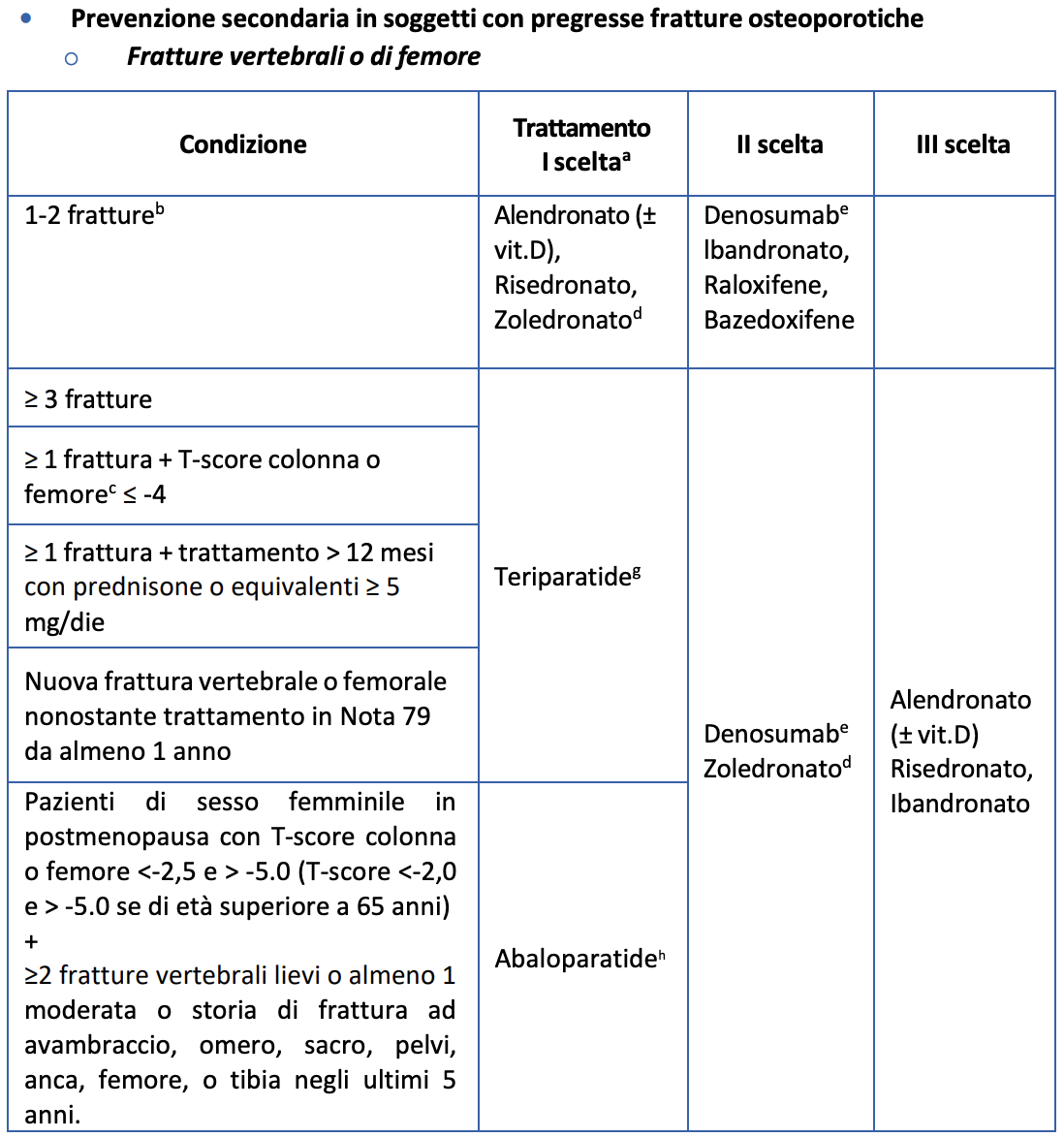
Nei soggetti con storia di frattura vertebrale o di femore:
• sesso femminile con osteoporosi documentata [laddove per osteoporosi documentata si intende: T-score colonna o femore < -2.5 (o < -2 se ≥ 2 fratture vertebrali moderate o gravi o frattura femorale negli ultimi due anni)];
• + anamnesi di ≥ 1 frattura vertebrale moderata o grave oppure ≥ 2 fratture vertebrali lievi oppure una frattura femorale nei due anni prima;
• + rischio di frattura a dieci anni ≥ 20% (determinato con calcolatore validato);
• + impossibilità a proseguire trattamenti alternativi efficaci (intolleranza, inefficacia o scadenza del periodo di impiego autorizzato);
• + assenza di pregressi eventi cardio- e cerebro-vascolari o presenza di rischio cardio-vascolare (CV) elevato (definito come rischio ≥ 20% secondo le carte del rischio del progetto CUORE).
Nei soggetti con pregressa frattura “non vertebrale e non femorale”:
• sesso femminile con T-score colonna o femore < -2.5;
• + anamnesi ≥ 2 fratture non vertebrali;
• + rischio di frattura a dieci anni ≥ 20% (determinato con calcolatore validato);
• + impossibilità a proseguire trattamenti alternativi efficaci (intolleranza, inefficacia o scadenza del periodo di impiego autorizzato);
• + assenza di pregressi eventi cardio- e cerebro-vascolari o presenza di rischio CV elevato (definito come rischio ≥ 20% secondo le carte del rischio del progetto CUORE).
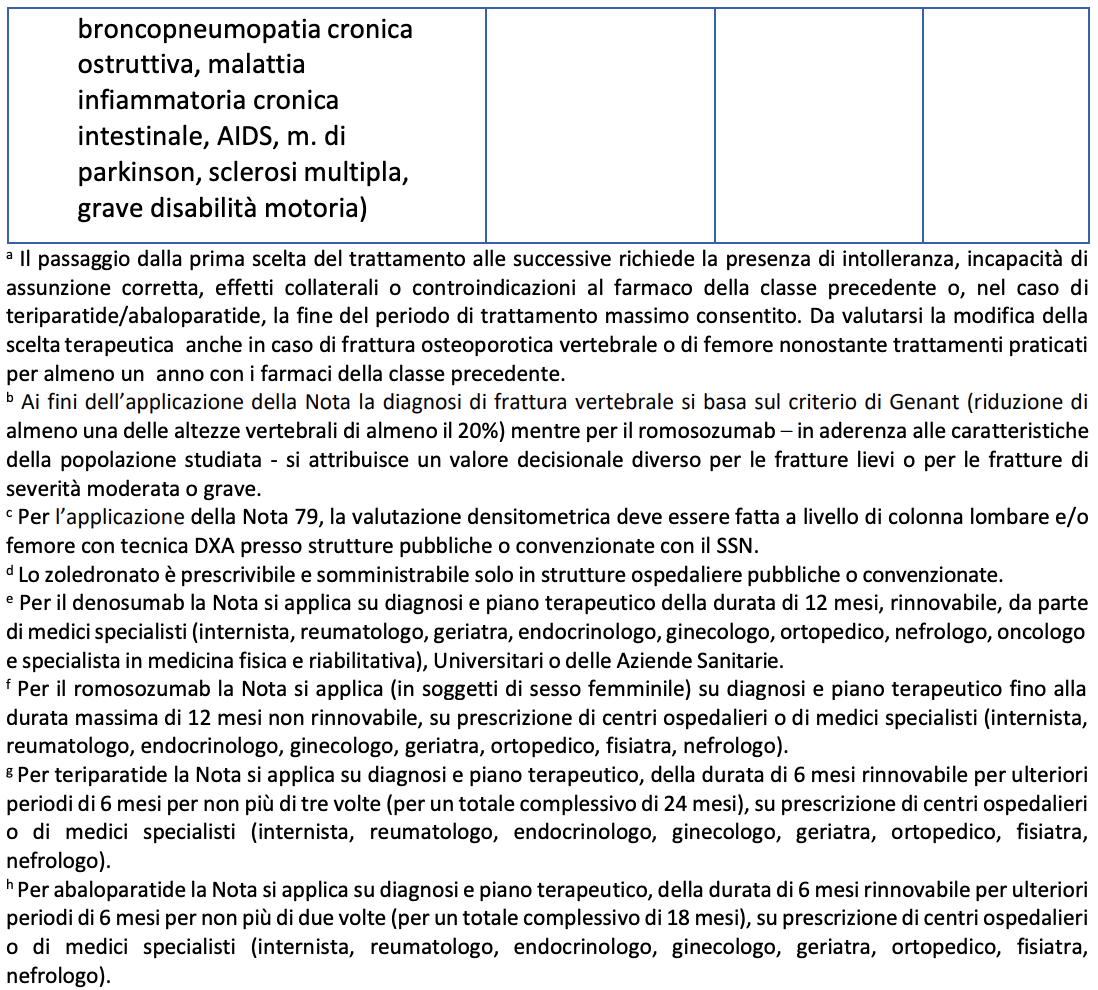
La storia di pregresso infarto e/o ictus è, infatti, una controindicazione alla terapia. Il piano terapeutico web based, come specificato nella nota, prevede:
• nei soggetti di età < 70 anni l’utilizzo delle carte per il rischio CV;
• nei pazienti ≥ 70 anni, per i quali non possono essere usate le carte italiane del Progetto Cuore, la presenza o meno di fattori di rischio CV (storia di accertata patologia CV, ipertensione arteriosa, iperlipidemia,
diabete mellito, tabagismo, insufficienza renale grave ed età) deve essere attentamente vagliata nella scelta di sottoporre un paziente a trattamento con romosozumab.


CONTROINDICAZIONI
- IPERSENSIBILITA’ al principio attivo o ad uno qualsiasi degli eccipienti
- IPOCALCEMIA: in pazienti trattate con romosozumab sono stati riscontrati casi di ipocalcemia transitoria. L’ipocalcemia deve essere corretta prima di iniziare la terapia e le pazienti devono essere monitorate per lo sviluppo di segni e sintomi di ipocalcemia. Se una paziente presenta sintomi sospetti di ipocalcemia durante il trattamento, occorre valutare i livelli di calcio. Le pazienti devono assumere un’adeguata integrazione di calcio e vitamina D. Le pazienti con compromissione renale severa (velocità di filtrazione glomerulare stimata [eGFR] 15- 29 ml/min/1,73 m2) o sottoposte a dialisi sono a maggior rischio di sviluppare ipocalcemia ed i dati di sicurezza per queste pazienti sono limitati. In queste pazienti, i livelli di calcio devono essere monitorati.
- Anamnesi di INFARTO MIOCARDICO o ICTUS o rischio cardio-vascolare elevato (≥20%): nonostante le premesse cliniche incoraggianti, il riscontro di un incremento non spiegato del rischio di eventi cerebrocardiovascolari, ha condotto a limitare prudenzialmente l’impiego del romosozumab escludendo i soggetti con pregressi eventi cerebrocardiovascolari o con condizioni di rischio cardiovascolare. Se una paziente manifesta infarto miocardico o ictus durante la terapia, il trattamento con romosozumab deve essere interrotto.
- GRAVIDANZA E ALLATTAMENTO
PARTICOLARI AVVERTENZE
- ANZIANI: nelle pazienti anziane non è necessario alcun aggiustamento della dose.
- INSUFFICIENZA RENALE: per le pazienti con compromissione renale non è richiesto alcun aggiustamento della dose. Nelle pazienti con compromissione renale severa o sottoposte a dialisi occorre monitorare i livelli sierici di calcio.
- FRATTURE ATIPICHE DEL FEMORE: in pazienti trattate con romosozumab è stata riportata raramente la frattura atipica a bassa energia o da trauma lieve della diafisi femorale, che può verificarsi spontaneamente. Per qualsiasi paziente che manifesti dolore di nuova insorgenza o insolito a livello della coscia, dell’anca o dell’inguine bisogna sospettare una possibile frattura atipica e la paziente deve essere valutata per escludere una frattura femorale incompleta. Le pazienti che presentano una frattura femorale atipica devono essere valutate anche per eventuali segni e sintomi di frattura nell’arto controlaterale. Deve essere considerata l’interruzione della terapia con romosozumab, in base a una valutazione individuale del rapporto beneficio/rischio..
- OSTEONECROSI DELLA MASCELLA/MANDIBOLA (ONJ): è stata riportata raramente nelle pazienti trattate con romosozumab. I seguenti fattori di rischio devono essere considerati quando si valuta il rischio di una paziente di sviluppare ONJ:
- la potenza del medicinale che inibisce il riassorbimento osseo (il rischio aumenta con la potenza anti-riassorbitiva del composto) e la dose cumulativa della terapia anti-riassorbitiva stessa;
- tumore, condizioni di comorbilità (es. anemia, coagulopatie, infezione), fumo;
- terapie concomitanti: corticosteroidi, chemioterapia, inibitori dell’angiogenesi, radioterapia della regione testa-collo;
- scarsa igiene orale, malattia parodontale, protesi dentarie non inserite correttamente, anamnesi di malattia dentaria, procedure dentarie invasive, es. estrazioni dentarie.
Tutte le pazienti devono essere incoraggiate a mantenere una buona igiene orale, a sottoporsi a controlli dentali periodici e segnalare immediatamente qualsiasi sintomo a livello orale come mobilità dentale, dolore o gonfiore o la mancata guarigione di ulcere o perdite durante il trattamento con romosozumab. Le pazienti che si sospetta presentino ONJ o che sviluppano ONJ mentre assumono romosozumab devono ricorrere alle cure di un dentista o rivolgersi a un chirurgo del cavo orale esperto di ONJ. È necessario valutare l’interruzione della terapia con romosozumab fino a quando la condizione non si sarà risolta e i fattori di rischio che hanno contribuito al suo insorgere saranno stati mitigati, ove possibile.
INTERAZIONI
Non sono stati effettuati studi d’interazione farmacologica con romosozumab. Non si prevedono interazioni farmacocinetiche con romosozumab.
E NEL MASCHIO?
Nel maschio l’efficacia terapeutica è stata valutata in trials controllati e randomizzati per alendronato, risedronato, zoledronato, denosumab-e romosozumab. Il numero dei pazienti studiati nei trials era modesto e non era calcolato per valutare gli effetti del trattamento sulle fratture osteoporotiche. L’efficacia per la prevenzione delle fratture è quindi in parte surrogata dai dati sulla massa ossea e non è al momento riconosciuta per il romosozumab.
EFFETTI FARMACODINAMICI
Nelle donne in post-menopausa con osteoporosi, romosozumab ha aumentato i livelli del marcatore di formazione ossea pro-peptide N-terminale del pro-collagene di tipo 1 (P1NP) nelle fasi precoci del trattamento, con un picco di incremento pari a circa il 145% rispetto al placebo 2 settimane dopo l’inizio del trattamento, seguito da un ritorno ai livelli del placebo al mese 9 e una riduzione a circa il 15% al di sotto del livello del placebo al mese 12. Romosozumab ha ridotto i livelli del marcatore di riassorbimento osseo telopeptide C-terminale del collagene di tipo 1 (CTX) con una riduzione massima di circa il 55% rispetto al placebo 2 settimane dopo l’inizio del trattamento. I livelli di CTX si sono mantenuti al di sotto del placebo e al mese 12 erano di circa il 25% inferiori rispetto al placebo.
Dopo l’interruzione della terapia con romosozumab nelle donne in post-menopausa con osteoporosi, i livelli di P1NP sono ritornati al valore basale entro 12 mesi; i livelli di CTX sono aumentati oltre i livelli basali entro 3 mesi e ritornati verso valori basali entro il mese 12, riflettendo la reversibilità dell’effetto. Una volta ripreso il trattamento con romosozumab (in un numero limitato di pazienti) dopo 12 mesi di trattamento con placebo, l’aumento di P1NP e la riduzione di CTX indotti da romosozumab presentavano livelli simili a quelli osservati durante il trattamento iniziale.
EFFETTI FARMACOCINETICI
- ASSORBIMENTO: il tempo mediano al raggiungimento della concentrazione massima di romosozumab (tmax) era di 5 giorni (intervallo: 2-7 giorni). Dopo una dose sottocutanea di 210 mg, la biodisponibilità era dell’81%.
- BIOTRASFORMAZIONE: essendo un anticorpo monoclonale umanizzato (IgG2) con elevata affinità e specificità per la sclerostina, pertanto viene eliminato attraverso una via di eliminazione rapida saturabile (ovvero, clearance non lineare mediata dal target, mediata dalla degradazione del complesso romosozumab- sclerostina) e attraverso una via di eliminazione lenta non specifica mediata dal sistema reticoloendoteliale.
- ELIMINAZIONE: una volta raggiunta la Cmax, i livelli sierici sono diminuiti con un’emivita media di 12,8 giorni. Lo stato stazionario è stato raggiunto generalmente entro il 3° mese con un accumulo inferiore a 2 volte in seguito a somministrazione mensile.
- COMPROMISSIONE RENALE: dopo somministrazione di una dose di romosozumab di 210 mg in una sperimentazione clinica su 16 pazienti con compromissione renale severa (clearance della creatinina < 30 ml/min) o nefropatia allo stadio terminale (ESRD) sottoposti a emodialisi, la Cmax e l’AUC (area sotto la curva) medie erano del 29% e 44% più alte nei pazienti con compromissione renale severa rispetto ai soggetti sani. L’esposizione media a romosozumab era simile nei pazienti con ESRD sottoposti a emodialisi rispetto ai soggetti sani. L’analisi farmacocinetica di popolazione indicava un aumento nell’esposizione a romosozumab proporzionale alla gravità della compromissione renale. Tuttavia, in base a un modello di esposizione- risposta delle variazioni nella BMD e al confronto con le esposizioni ottenute a dosi cliniche tollerate, non sono raccomandati aggiustamenti della dose in questi pazienti. Nelle pazienti con compromissione renale severa o sottoposte a dialisi si raccomanda il monitoraggio dell’ipocalcemia
- COMPROMISSIONE EPATICA: non sono state condotte sperimentazioni cliniche per valutare l’effetto della compromissione epatica. La compromissione epatica non dovrebbe influire sulla farmacocinetica di romosozumab in quanto il fegato non è l’organo maggiormente coinvolto nel metabolismo o nell’escrezione di romosozumab.
- ANZIANI: la farmacocinetica di romosozumab non è risultata influenzata dall’età nell’intervallo compreso tra 20 e 89 anni.
- PESO CORPOREO: l’esposizione a romosozumab è diminuita con l’aumento del peso corporeo; tuttavia, tale diminuzione ha avuto un impatto minimo sull’aumento della BMD a livello della colonna lombare in base all’analisi di esposizione-risposta e non è clinicamente significativo. In base all’analisi farmacocinetica di popolazione, la previsione per l’AUC mediana dello stato stazionario per una paziente di 61 kg e una di 114 kg è rispettivamente di 558 µg/die/ml e di 276 µg/die/ml in seguito a una dose mensile di 210 mg di romosozumab.
- ETNIA E SESSO: non sono necessari aggiustamenti della dose per alcuna caratteristica della paziente. In base a un’analisi farmacocinetica di popolazione, sesso e razza (giapponese vs non giapponese) non hanno avuto alcun impatto clinicamente significativo sulla farmacocinetica di romosozumab (variazione < 20% nell’esposizione allo stato stazionario).
E DOPO IL ROMOSOZUMAB?
Nelle donne in postmenopausa con osteoporosi che hanno completato un ciclo di romosozumab, si raccomanda il trattamento con terapie antiriassorbitive (bisfosfonati, denosumab) per mantenere i guadagni di densità minerale ossea e ridurre il rischio di frattura (Shoback, Dolores et al. “Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society Guideline Update.” The Journal of clinical endocrinology and metabolism vol. 105,3 (2020): dgaa048. doi:10.1210/clinem/dgaa048)
FONTI
- https://www.gazzettaufficiale.it/eli/gu/2022/08/29/201/sg/pdf
- https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_000747_048408_FI.pdf&retry=0&sys=m0b1l3
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4824823/pdf/223_2016_Article_126.pdf
- Shoback, Dolores et al. “Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society Guideline Update.” The Journal of clinical endocrinology and metabolism vol. 105,3 (2020): dgaa048. doi:10.1210/clinem/dgaa048

Lascia un commento